Mastering Reaction Pathways: A Deep Dive into Sn2, Sn1, E1, and E2 Mechanisms Using the Advanced Reaction Chart
David Miller
3494 views
Mastering Reaction Pathways: A Deep Dive into Sn2, Sn1, E1, and E2 Mechanisms Using the Advanced Reaction Chart
At the heart of organic chemistry lies the intricate dance of molecular transformations governed by reaction mechanisms—each pathway governed by subtle electronic and structural influences. Understanding how substitution and elimination reactions proceed—Sn2, Sn1, E1, and E2—requires more than memorization; it demands a precise visual framework that maps reactivity, conditions, and outcomes. The Sn2, Sn1, E1, E2 Chart is a predictive compass that decodes these dynamic choices, revealing the factors that steer a reaction toward substitution or elimination.
Drawing on fundamental principles and modern examples, this analysis explores how these mechanisms interconnect, their defining characteristics, and how chemists leverage them in synthesis and process design. The chart’s role as a decision-making tool is indispensable, transforming abstract behavior into actionable insight.
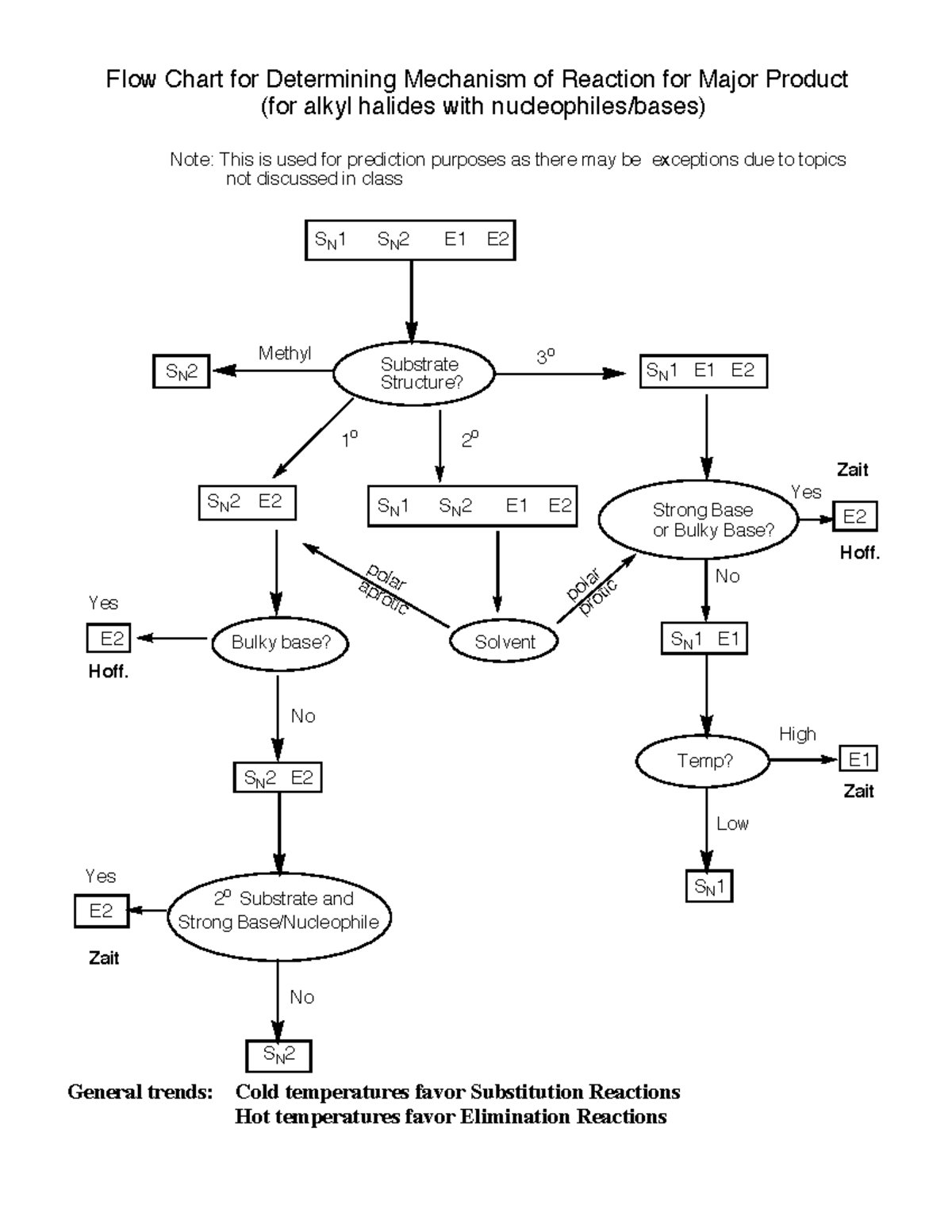
At the core of organic mechanism selection lies the interplay between carbon-substrate structure, nucleophile/base strength, solvent polarity, and temperature—variables tightly mapped in the Sn2, Sn1, E1, E2 Chart.
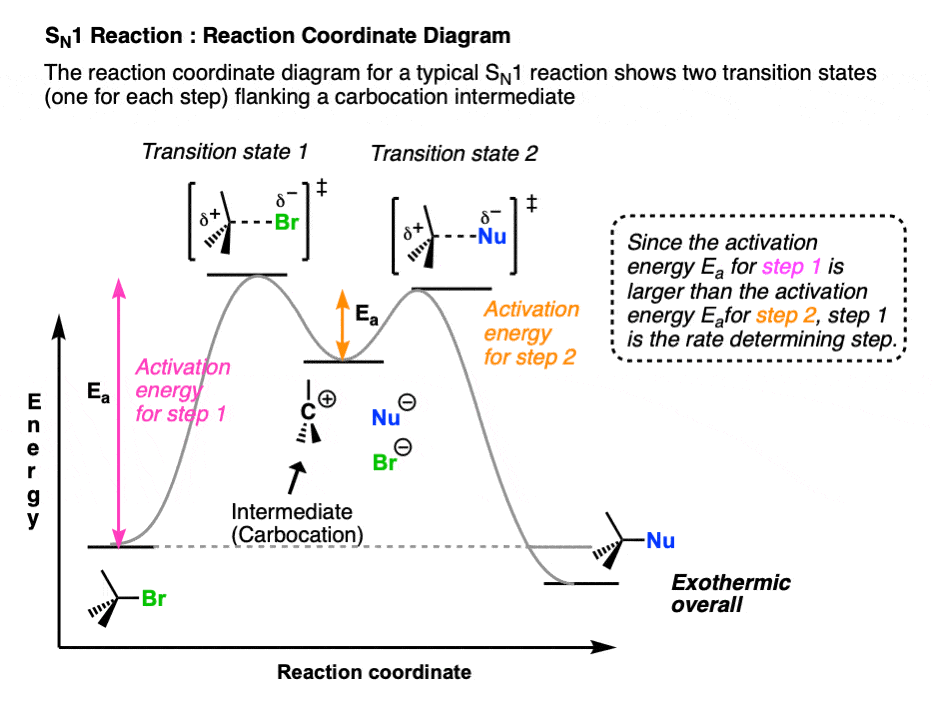
This chart does not merely list reactions; it illustrates how conditions alter mechanistic pathways, dictating whether a bimolecular nucleophilic substitution (Sn2) dominates, or a unimolecular elimination (E2) or dissociative process (Sn1/E1) prevails. For instance, Sn2 reactions thrive with strong nucleophiles and polar aprotic solvents, favoring primary substrates due to reduced steric hindrance—allowing a seamless backside attack (Figure 1). In contrast, Sn1 processes rely on carbocation stability, making tertiary substrates ideal for solvolysis under mild conditions, where solvent molecules stabilize the evolving positive charge (Figure 1).
E1 and E2 eliminations follow similar logic but hinge on base strength and proton availability, with E2 demanding precise alignment of antiperiplanar geometry. Navigating these choices with clarity demands the chart as a guidepost.
Sn2 Mechanism: A Bimolecular Dance of Precision
The Sn2 mechanism, standing for bimolecular nucleophilic substitution, is defined by its single-step rate-determining process where nucleophile and substrate collide in a backside attack, displacing a leaving group bidirectionally. This concerted transformation results in inversion of stereochemistry at the reaction center—a signature hallmark visible in chiral substrates like 2-chlorobutane with a nucleophile such as sodium iodide in DMSO.
The reaction rate depends on the concentration of both nucleophile and substrate (second-order kinetics), making it highly sensitive to steric crowding.
Critical conditions for Sn2 include: - A strong nucleophile (e.g., hydroxide, cyanide, alkoxides) with high charge density - A polar aprotic solvent (e.g., acetone, DMF), which solvates cations but not the nucleophile, enhancing reactivity - Primary and secondary alkyl halides being most reactive; tertiary substrates are effectively inert due to steric blockage (Figure 1) The stereochemical outcome is unambiguous: Richard Wilstrop’s classic inversion example demonstrates how a wedge (bond) flips to a dash upon nucleophilic assault, preserving absolute configuration only when a mirror image forms—key for synthetic stereocontrol. “Because the bond formation and breaking are simultaneous,” notes organic chemist Dr.
Elena Marquez, “Sn2 reactions offer unparalleled selectivity in complex molecules.” When applied strategically, Sn2 enables efficient carbon-carbon and carbon-heteroatom bond formation, especially in pharmaceuticals where precise stereochemistry is non-negotiable.
Sn1 Mechanism: A Two-Step Pathway to Carbocation Formation
In contrast, the Sn1 mechanism—unimolecular nucleophilic substitution—proceeds through a two-step process: first, the leaving group departs to form a carbocation intermediate, followed by nucleophilic attack. Second-order rate dependence reflects reliance on substrate concentration alone, making it favored by tertiary substrates with electron-donating alkyl groups that stabilize positive charge development. Poor leaving groups and polar protic solvents (e.g., water, alcohols) further promote Sn1 by solvating both the leaving group and developing carbocation, lowering activation energy.
Key conditions promoting Sn1 include: - Tertiary, secondary, or less substituted sp³ halides with weakly basic leaving groups (e.g., iodide, tosylate) - Polar protic solvents that stabilize ions via hydrogen bonding - Slow, mild nucleophilic attacks; strong bases may favor elimination instead The carbocation intermediate confers potential for rearrangement—hydride or alkyl shifts alter product distribution, adding complexity. “The elegance of Sn1 is its ability to form new stereochemistry—though with racemic mixtures unless stereochemistry is constrained,” explains Dr. Marquez.
This mechanism is pivotal in industrial syntheses, such as the industrial synthesis of ibuprofen, where abundant tertiary substrates ensure high yields via carbocation intermediates. Yet, its tendency to produce mixtures limits use when stereospecificity is required.
E1 and E1 Reactions: Hydroxy Elimination as a Solvent-Dependent Pathway
The E1 mechanism, or unimolecular elimination, shares with Sn1 the formation of a carbocation intermediate but diverges in kinetics and product outcome: it proceeds via a two-step sequence (substrate loss → carbocation → base-enhanced proton abstraction), with first-order rate dependence.
E1 elimination is favored under milder, temperature-driven conditions where solvent polarity enables ion separation without strong nucleophilic demand. Though less common than Sn1 in synthetic chemistry, E1 plays an underappreciated role—particularly in soft solvent environments where nucleophile-base competition tilts the balance toward elimination.
Where E1 and Sn1 overlap, E2 reactions present a starkly different mechanism: a single concerted step where base abstracts a β-hydrogen antiperiplanar to the leaving group, facilitating simultaneous Brønsted acid-like proton removal and bond rupture to form alkene.
Unlike E1, E2 demands a strong base (e.g., hydroxide, alkoxide) and efficient orbital alignment, yielding regio- and stereospecific alkenes per Zaitsev’s rule—where energy favors the more substituted product.
Critical distinctions emerge: - E1/E2: old, less electron-demanding elimination, often via Sn1 intermediates or direct abstraction - E2: modern, base-programmed, reliant on antiperiplanar geometry and base strength “E2 dominates in competitive systems where the base is strong enough to force simultaneous departure,” notes reaction dynamics expert Dr. Alan Ruiz.
“But E1 offers cost-effective routes when nothing beyond solvent or base is available—think waste-stream processing or closed-loop reactions.” The E2 pathway’s stereospecificity aligns with precise product filtering, essential in fine chemical synthesis, allowing chemists to tailor alkenes with confidence. Still, overworking E1 conditions risks competing Sn1 processes, underscoring the need for precise parameter control.